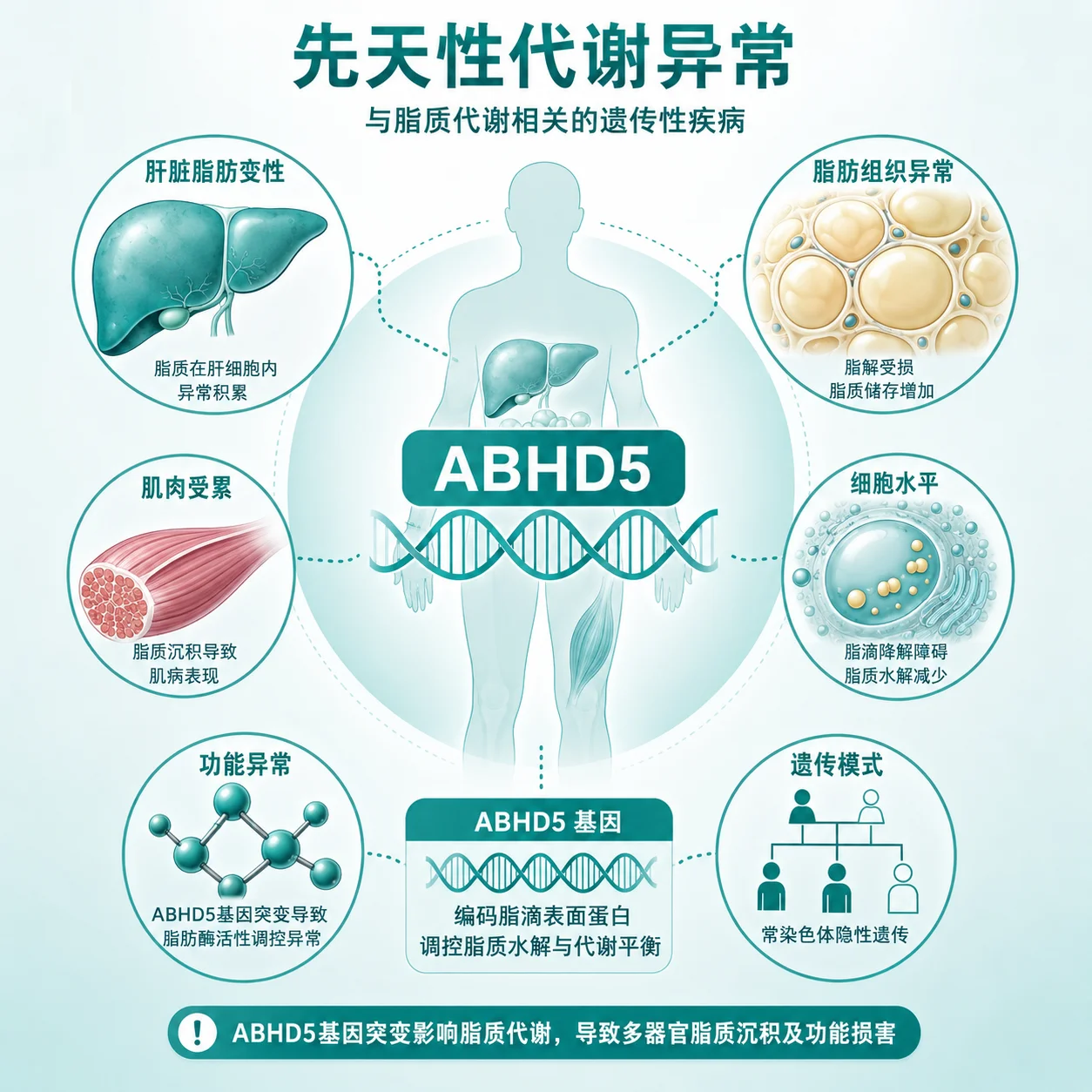
什么是Chanarin-Dorfman 综合征?
Chanarin-Dorfman综合征是一种罕见的遗传代谢病,它影响身体对脂肪的正常处理和储存。简单来说,患者的细胞里缺少一种关键的酶,导致从食物中摄取或身体自身合成的脂肪(主要是甘油三酯)无法被有效分解,从而异常地堆积在全身各种细胞中,尤其是皮肤、肝脏、肌肉和血液细胞里。这种脂肪的异常累积是引发后续一系列症状的根本原因。
临床表现有哪些?
这种综合征的症状表现多样,且可能在不同患者身上有轻有重。最常被注意到的往往是皮肤的改变,表现为出生后不久出现的鱼鳞病样皮损,皮肤干燥、增厚,伴有细小的鳞屑。除了皮肤,内脏器官也会受累,很多患者会出现肝脏和脾脏的肿大。在血液检查中,医生可能会发现一种被称为“Jordan异常”的现象,这指的是在显微镜下看到中性粒细胞(一种白细胞)的细胞质里存在脂质空泡。此外,部分患者可能伴有肌肉无力或肌病的表现。这些症状通常都是逐渐发展的。
这个病会遗传吗?
Chanarin-Dorfman综合征遵循常染色体隐性遗传方式。这意味着,只有当一个人从父母双方各继承了一个有缺陷的ABHD5等基因拷贝时,才会发病。如果一个人只携带了一个有缺陷的基因拷贝,他/她通常不会表现出疾病症状,被称为携带者。当父母双方都是携带者时,他们每次生育,孩子有25%的概率患病,50%的概率成为像父母一样的健康携带者,还有25%的概率完全健康且不携带缺陷基因。了解这一遗传模式,有助于理解家族内的发病情况。
怎么确诊?
对于这类临床表现复杂且罕见的疾病,诊断需要综合评估。医生通常会从详细的病史询问和体格检查开始,特别是关注皮肤、肝脾和肌肉的状况。血液检查,尤其是血涂片镜检寻找粒细胞中的脂质空泡(Jordan异常),是一个重要的线索。为了明确诊断,此类疾病通常通过基因检测辅助诊断,寻找ABHD5等基因是否存在致病变异。基因检测的结果需要由临床医生结合患者的具体表现进行综合解读。如有相关表现,建议就医由专科医生评估。
哪些基因相关?
目前已知的致病基因主要是ABHD5基因。这个基因负责指导合成一种对脂肪分解至关重要的酶。正常情况下,这种酶能激活另一种脂肪分解酶,共同将细胞内的甘油三酯分解为脂肪酸供能。当ABHD5基因发生致病性变异时,酶的功能丧失或严重受损,脂肪分解的链条在起始环节就中断了,导致甘油三酯在细胞内不断堆积。基因检测的意义在于确认这种分子层面的缺陷,为临床诊断提供关键依据,并帮助明确遗传模式。
目前怎么治疗?预后如何?
目前对于Chanarin-Dorfman综合征尚无根治性的治疗方法,临床管理主要以对症和支持治疗为主。针对鱼鳞病,可以使用润肤剂和外用药来改善皮肤状况。对于肝脏脂肪变性和肿大,需要定期监测肝功能,并采取保护肝脏的生活方式。肌肉症状也可能需要相应的康复管理。疾病的长期预后存在个体差异,部分患者可能病情相对稳定,而严重累及肝脏者可能需要更密切的医疗关注。整体的治疗策略需要根据患者的具体情况由多学科团队制定。
能预防吗?家人需要筛查吗?
对于已有患病成员的家庭,遗传咨询非常重要。通过基因检测明确先证者(首个被诊断的家庭成员)的致病基因变异后,可以对其他家庭成员进行携带者筛查。对于有再生育计划的携带者夫妇,可以在专业指导下了解产前诊断或胚胎植入前遗传学检测等选项的可能性,这些技术旨在帮助评估胎儿是否患病。这是一种主动了解遗传风险、为家庭生育计划提供信息支持的方式。如有相关表现,建议就医由专科医生评估。