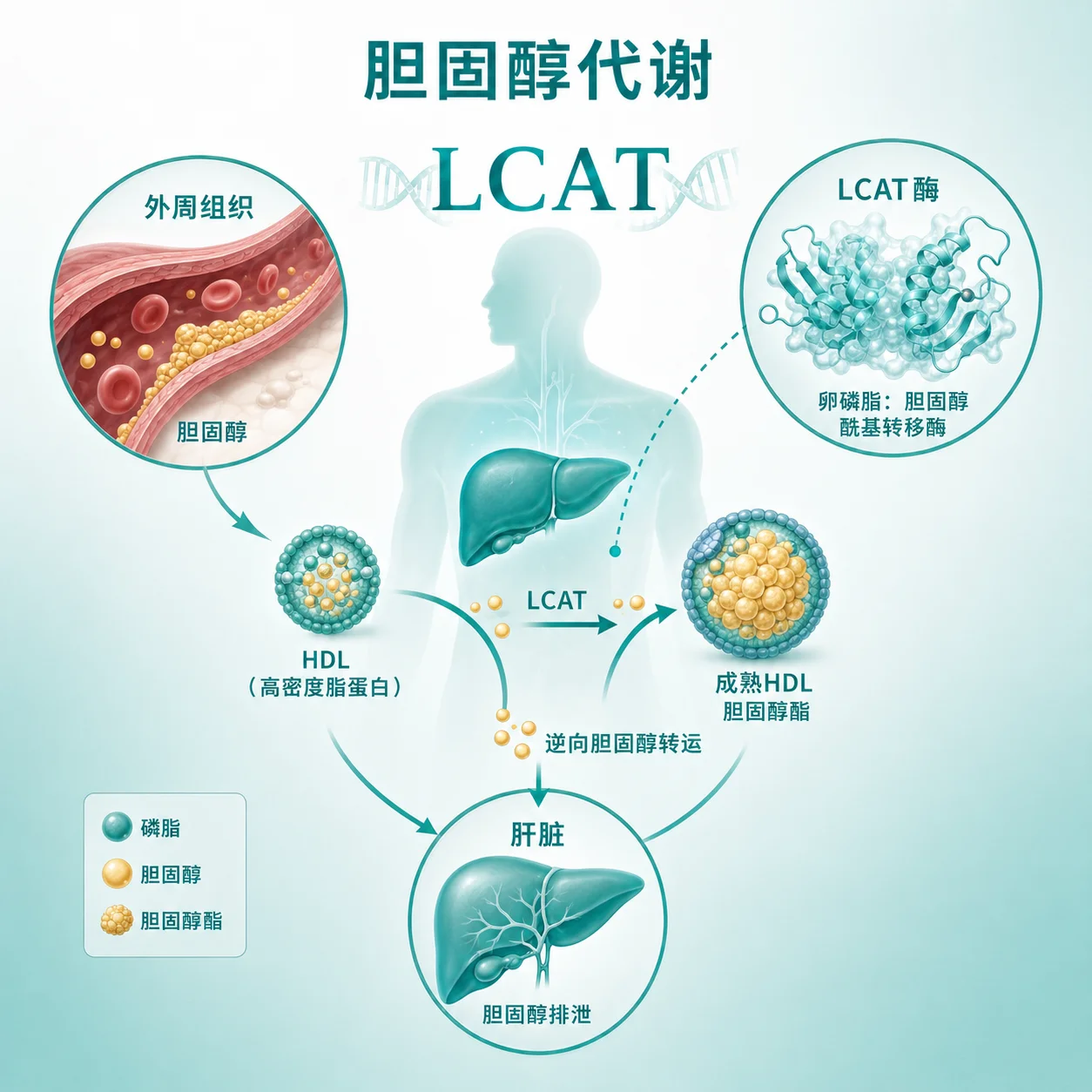
什么是家族性卵磷脂胆固醇酞基转移酶缺乏症?
家族性卵磷脂胆固醇酰基转移酶缺乏症是一种罕见的遗传性疾病,它源于体内一种重要的酶——卵磷脂胆固醇酰基转移酶(LCAT)的功能缺失或严重不足。这种酶在胆固醇代谢过程中扮演着关键角色,负责将胆固醇与卵磷脂结合,形成更稳定的酯化胆固醇。当这种酶缺乏时,胆固醇的正常转运和代谢就会受到影响,导致一系列身体变化。
临床表现有哪些?
这种疾病的表现可能因人而异,但常见的观察包括眼睛角膜出现混浊,这可能会影响视力清晰度。血液方面可能出现溶血性贫血,即红细胞过早破坏导致贫血相关表现。肾脏也可能受到影响,出现蛋白尿,长期可能发展为肾病。此外,血液检查常会发现高密度脂蛋白胆固醇水平极低。这些症状通常在儿童或青少年时期开始显现,并可能随时间进展。如有相关表现,建议就医由专科医生评估。
这个病会遗传吗?
这种疾病的遗传方式是常染色体隐性遗传。这意味着一个人需要从父母双方各获得一个异常的基因拷贝才会患病。如果只从一方获得一个异常拷贝,则成为携带者,通常不会表现出疾病症状,但可能将异常基因传给下一代。对于患病者的子女,如果配偶是携带者或患者,则子女有一定患病风险;如果配偶不携带异常基因,则子女会成为携带者但不患病。了解家族遗传模式有助于家庭成员理解潜在风险。
怎么确诊?
此类疾病通常通过基因检测辅助诊断,结合临床表现和实验室检查结果进行综合判断。诊断过程一般包括详细的病史询问、体格检查、血液化验(如血脂分析、肾功能检查)以及眼科评估。当怀疑此类遗传性疾病时,基因检测可以帮助确认是否存在LCAT基因的特定变异。诊断需要由专科医生在全面评估后做出。
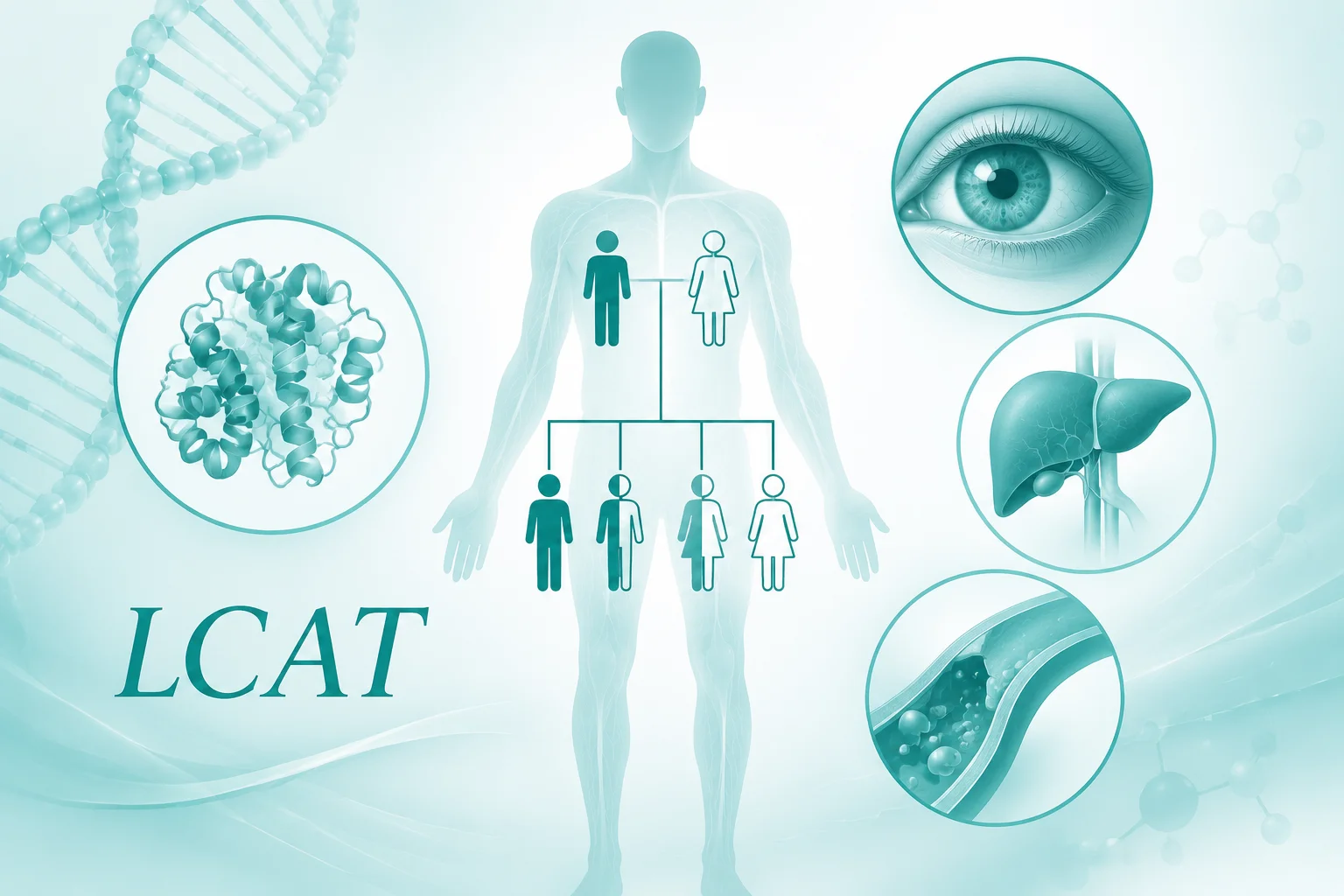
哪些基因相关?

LCAT基因负责编码卵磷脂胆固醇酰基转移酶,这种酶的功能是将游离胆固醇转化为酯化胆固醇,这是胆固醇正常运输和代谢的关键步骤。当该基因发生致病性变异时,酶的活性会显著降低或完全丧失,导致胆固醇代谢紊乱。基因检测的意义在于帮助确认病因,理解疾病的分子基础,并为家族成员提供遗传信息参考。检测结果应由专业医生结合临床情况解读。
目前怎么治疗?预后如何?
目前对于这种疾病尚无特效根治方法,治疗主要侧重于管理症状和预防并发症。例如,针对肾脏问题可能需要进行肾功能保护管理,贫血可能需要相应支持,眼科问题可能需要定期监测和干预。预后因人而异,取决于症状的严重程度和并发症的发展情况。一些患者可能在长期管理下保持相对稳定的状态,而另一些可能面临更多挑战。定期随访和专科护理对于维持健康很重要。
能预防吗?家人需要筛查吗?
作为一种遗传性疾病,它无法通过常规生活方式改变来预防。对于已知患病或携带异常基因的家庭,可以在专业遗传咨询指导下考虑产前诊断或家系筛查。产前诊断是指在孕期通过特定方法了解胎儿是否携带致病基因组合。家系筛查是指对家族成员进行基因检测,以明确携带者状态。这些选择涉及复杂的个人和家庭决策,应在充分了解信息和咨询专业医生后慎重考虑。