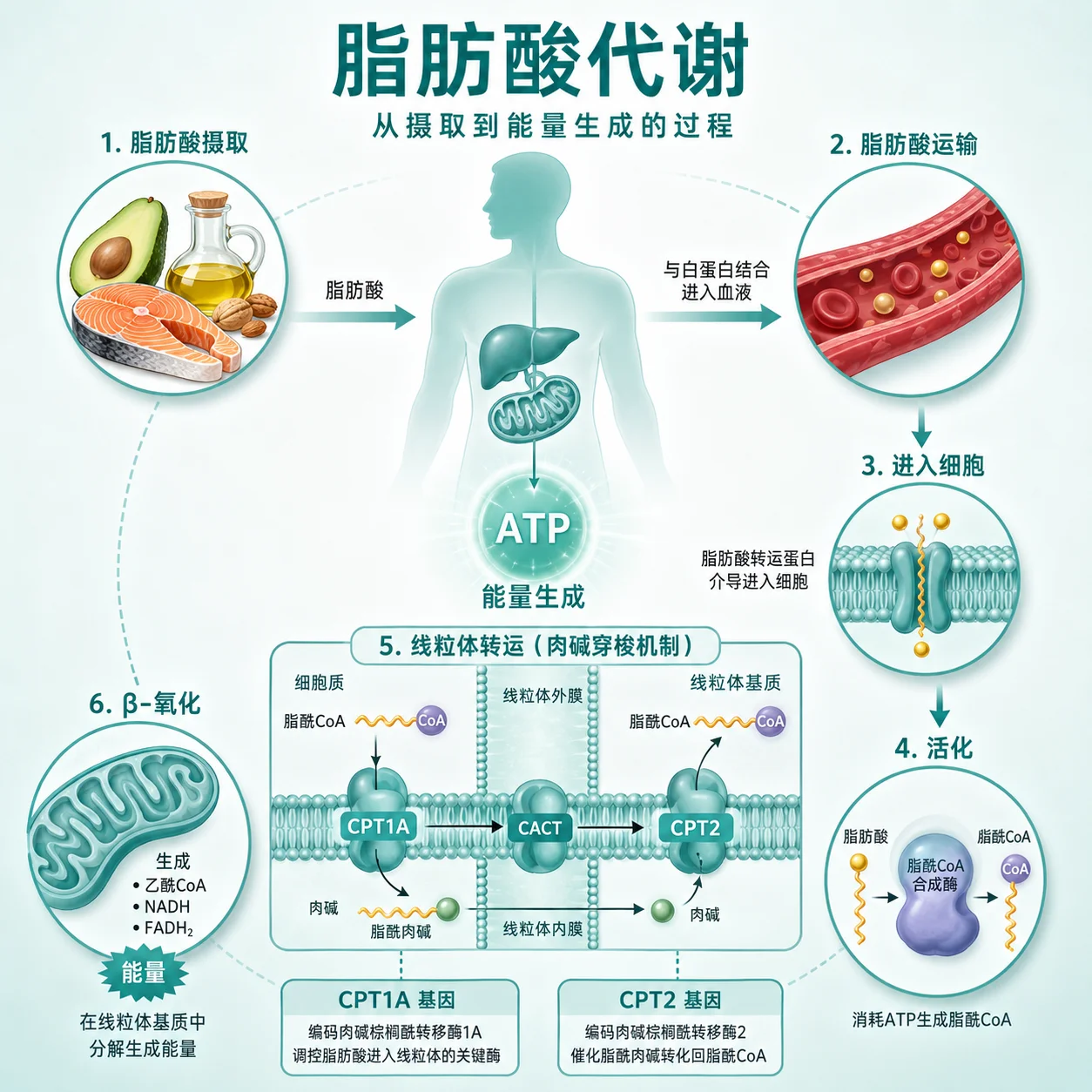
什么是肉碱棕榈酰转移酶I/ Ⅱ缺乏症?
肉碱棕榈酰转移酶I/II缺乏症是一种罕见的遗传代谢性疾病。它涉及身体利用脂肪作为能量来源的过程出现障碍。在正常情况下,肉碱棕榈酰转移酶像一座桥梁,帮助长链脂肪酸进入细胞内的能量工厂——线粒体,进行氧化分解产生能量。当这种酶的功能因基因变异而不足时,身体在需要大量能量(如长时间运动、饥饿或感染时)就无法有效利用脂肪储备,可能导致能量危机,影响多个器官系统。
临床表现有哪些?
临床表现因缺乏的酶类型和年龄而异。CPT I缺乏多见于婴儿期,主要表现为低酮性低血糖,即在血糖降低时,身体无法正常产生酮体作为替代能量,同时可能伴有肝脏肿大。CPT II缺乏则有两种常见形式:肌肉型通常在青少年或成人期显现,特点是运动后出现肌肉疼痛、无力,甚至可能发生横纹肌溶解,导致肌红蛋白尿;婴儿型则更为严重,可能早期就出现心肌受累、肝功能异常甚至衰竭。这些症状通常在能量需求增加的情况下诱发或加重。
这个病会遗传吗?
这种疾病遵循常染色体隐性遗传方式。这意味着一个人需要从父母双方各继承一个带有变异的基因拷贝才会患病。如果只从一方继承了一个变异拷贝,则成为携带者,通常不会表现出疾病症状,但可能将变异基因传递给下一代。对于已确诊患者的家庭,其子女有25%的概率患病,50%的概率成为无症状携带者,25%的概率完全健康。了解遗传模式有助于家庭成员理解风险,并在必要时进行遗传咨询。
怎么确诊?
此类疾病的诊断是一个综合过程。当医生基于临床表现怀疑此类代谢障碍时,通常会结合血液生化检查(如特定时段的血酮、肌酸激酶等)、影像学评估以及临床病史分析。基因检测在其中扮演重要角色,通过对CPT1A或CPT2等相关基因进行分析,可以确认是否存在致病性变异,从而辅助明确诊断。基因检测结果需要由专科医生结合临床表现进行综合解读。如有相关表现,建议就医由专科医生评估。
哪些基因相关?
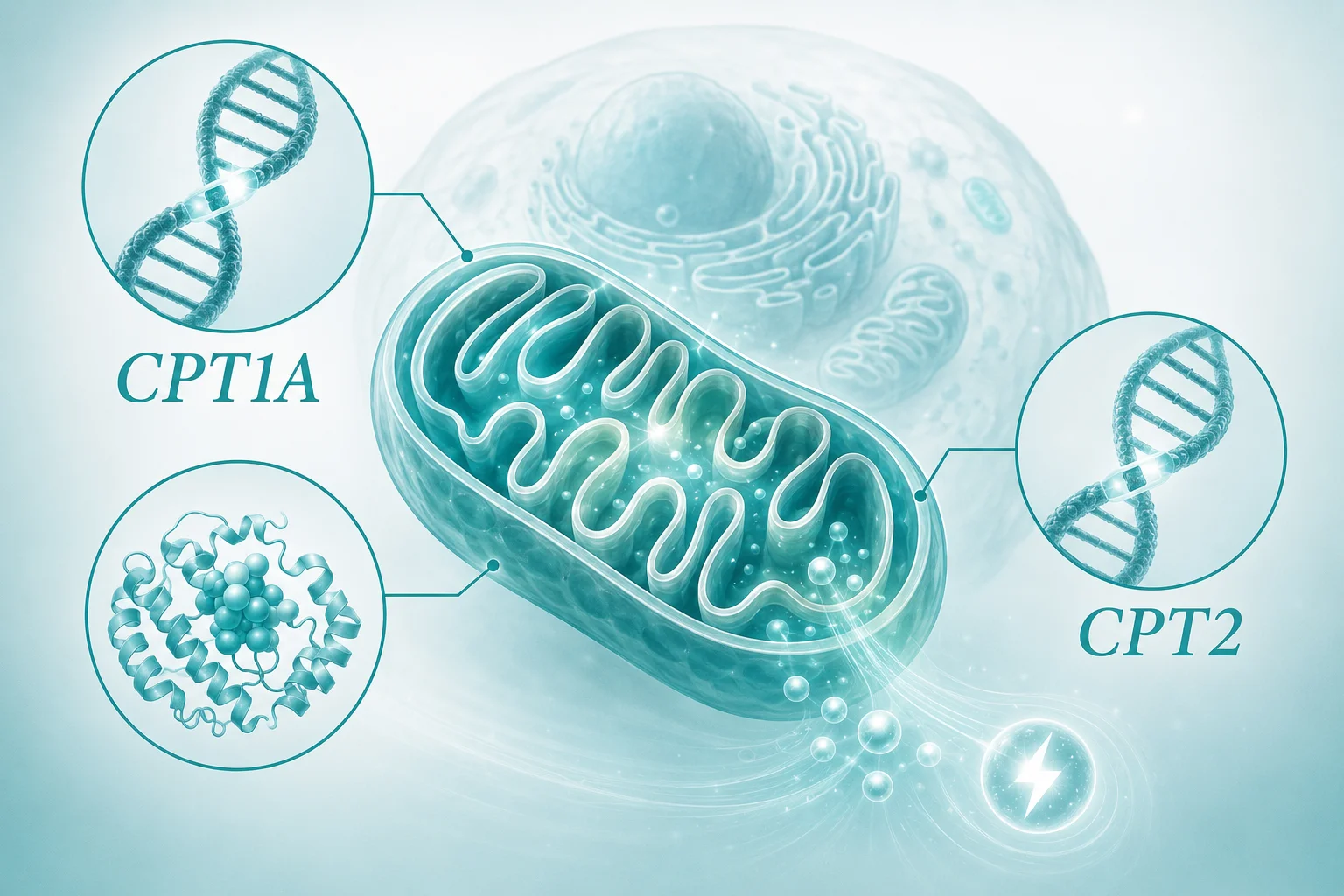
CPT1A和CPT2基因编码的蛋白质,正是肉碱棕榈酰转移酶I和II。CPT1酶主要负责将长链脂肪酸与肉碱结合,使其能够穿过线粒体外膜;CPT2酶则在脂肪酸进入线粒体基质后,将其从肉碱上解离,以便进行后续氧化。这两个基因的变异可能导致酶活性降低或丧失,阻断脂肪代谢通路。基因检测的意义在于识别这些变异,帮助解释疾病的生化基础,并为家庭提供明确的遗传信息。
目前怎么治疗?预后如何?
目前的治疗核心是预防能量危机事件的发生。管理策略包括避免长时间饥饿、保证规律进食,并在预期有高强度体力活动或感染时提前补充碳水化合物。对于肌肉型患者,调整运动强度和时间是关键。严重病例可能需要更密切的监测和支持性治疗。预后差异很大,早期识别、妥善管理以及避免诱发因素,对于改善长期健康状况非常重要。治疗需个体化,并在代谢专科医生指导下进行。
能预防吗?家人需要筛查吗?
对于已确诊家庭,预防措施主要围绕降低下一代患病风险。通过基因检测明确父母的基因状态后,可以进行遗传咨询。在知情选择的基础上,产前诊断(如通过绒毛取样或羊水穿刺获取胎儿细胞进行基因分析)是一种可选方案。此外,对家族中的其他成员进行携带者筛查,有助于了解各自的遗传风险。这些都属于预防性医学的范畴,需要在专业遗传咨询服务的框架下,根据家庭的具体情况和意愿来探讨和实施。